Berkeley Drosophila Genome Project
BDGP Resources
Around the Genomes
Genome Research 6: 71-79, 1996 The Drosophila Genome Project
Gerald M. Rubin Drosophila Genome Center, Department of Molecular and Cell Biology and Howard Hughes Medical Institute, Berkeley CA, 94720-3200
The Drosophila melanogaster genome is 165 Mb, with about 120 Mb of this being euchromatic. The genome is organized in 4 chromosome pairs (Figure 1A) and is estimated to contain 10 - 12,000 genes (G. Rubin and G. Miklos, unpubl.). The Drosophila research community-both through the efforts of "genome projects" and of individual investigators-has accumulated a vast amount of data on the genetic and molecular organization of the Drosophila genome as well as on the structure, expression and function of individual genes. In this review I will attempt to summarize the current state of genome research in Drosophila. The groups whose work is being summarized are listed in Table 1.
Table 1: Drosophila Research Community
| Group | Senior Members | Contacts | Major Funding Source |
| Berkeley Drosophila Genome Project (BDGP) | Drosophila Genome Center (DGC) Lawrence Berkeley National Lab. Human Genome Center (LBNL) Howard Hughes Medical Inst. (HHMI) |
http://www.fruitfly.org/ | NCHGR Department of Energy (DOE) |
| DGC | G. Rubin and S. Lewis (UC Berkeley) A. Spradling (Carnegie Inst. of Wash.) M. Palazzolo and C. Martin (LBNL) D. Hartl (Harvard) until 7/95. I Kiss (Szeged, Hungry) is a collaborator on the gene disruption project. |
http://www.fruitfly.org/ | NCHGR |
| LBNL | M. Narla, M. Palazzolo, C. Martin, J. Jaklevic and F. Eeckman | http://genome.lbl.gov | DOE |
| HHMI | G. Rubin and A. Spradling | HHMI | |
| European Drosophila Mapping Consortium | M. Ashburner (Cambridge) D. Glover and R. Saunders (Dundee) J. Modolell, CSIC (Madrid) F. Kafatos, EMBL (Heidelberg) K. Louis, B. Savakis, and I.Siden-Kiamos, IMBB (Heraklion) |
inga@nefelh.imbb.forth.gr | Fondation Schlumberger (Paris) MRC (UK) European Community Fundacion Ramon Areces |
| Karpen | G. Karpen, Salk Institute (La Jolla) | gary_karpen@qm.salk.edu | NCHGR |
| University of Alberta, Canada | J. Locke, A. Ahmed, J. Bell, H. Mc Dermid, D. Nash, D. Pilgrim, K. Roy and R. Hodgetts | rhodgett@pop.srv.ualberta.ca | Canadian Genome Analysis and Technology Program |
| McGill University, Canada | P. Lasko and B. Suter | Paul_Lasko@maclan.mcgill.ca | Canadian Genome Analysis and Technology Program |
| Duncan | I. Duncan (Washington Univ.) | Duncan@biodec.wustl.edu | NCHGR |
| FlyBase | W. Gelbart (Harvard); M. Ashburner (Cambridge, UK); T. Kaufman and K. Mathhews (Indiana Univ.) | http://morgan.harvard.edu/ flybase.bio.indiana.edu http://www.embl-ebi.ac.uk /flybase/ |
NCHGR MRC (UK) |
The origins of genome research in Drosophila Drosophila has been a leading organism for genome research for over 80 years. The concept that recombination frequencies could be used to order genes on a linear map was first demonstrated, and the first genetic maps were constructed, using Drosophila in 1913 (Sturtevant, 1913). Since then Drosophila has remained the metazoan with the most accurate and complete genetic map. The first physical maps-that is, maps that relate genetic functions to physical locations on chromosomes-were Bridges' polytene chromosome maps that, while made nearly 60 years ago (Bridges, 1937), had a resolution of +/- 100 kb. The polytene chromosome maps have allowed hundreds of genes to be placed in small physical intervals by classical cytogenetic methods. In the early 1970's when recombinant DNA methods were developed, Drosophila was the only organism with a physical map of its genome. This map, together with the sensitive and precise mapping that could be accomplished by in situ hybridization to polytene chromosomes (Pardue et al., 1970), enabled a number of pioneering studies to be carried out in David Hogness' laboratory in mid 1970's. Among these were the first chromosomal mapping of cloned unique and dispersed repetitive DNA segments (Wensink et al., 1974; Rubin et al., 1976) and the development of procedures for screening clones by colony hybridization, for assembling large chromosomal contigs, and for positional cloning (Grunstein and Hogness, 1975; Bender et al., 1979). Dozens of Drosophila genes that had been identified by mutations with interesting developmental or physiological phenotypes were positionally cloned in the early 1980s. Physical Maps The physical map based on the polytene chromosomes, while invaluable in providing an unambiguously correct reference map, is a cytogenetic map and thus can not meet the needs of the research community for a clone-based map. The first systematic attempts to make clone-based physical maps of entire Drosophila genome were the YAC-based maps of the Hartl and Duncan groups and the cosmid-based maps of the European Drosophila Mapping Consortium (EDMC). In 1992, the Berkeley Drosophila Genome Project (BDGP) began construction of a P1-based STS-content map. These maps are all cross-referenced to one another through the polytene chromosome map; moreover, the EDMC and BDGP maps contain additional cross-references through STSs. YAC-based Maps The YAC maps were constructed by carrying out in situ hybridization to polytene chromosomes with individual YACs. Hartl's group (Garza et al., 1989; Ajioka et al., 1991) mapped 1193 YAC clones with an average insert size of 207 kb and Duncan's group (Cai et al., 1994) mapped 855 euchromatic YACs with an average size of 211 kb. Together these YAC maps cover about 90% of the euchromatic genome for the autosomes, and 80% for the X; however, overlaps between YACs have not been confirmed by molecular methods. The distribution of clones appears to be essentially random over most of the euchromatic genome; however, there are a few regions for which no or very few YAC clones were recovered. Cosmid-based Maps The overall approach being used by the EDMC to construct a cosmid-based map consists of producing individual contig maps each representing a single chromosomal division (about 1 Mb), that are then complemented by the inclusion of STS markers, generated from the ends of the inserts of mapped cosmids (Siden-Kiamos et al., 1990). In brief, an arrayed cosmid library is screened with a probe generated by micro-dissection of polytene chromosomes and the DNA from positive clones is fingerprinted. After computer-assisted ordering of overlapping cosmids into contigs, a representative set of cosmids from each contig are mapped by in situ hybridization to polytene chromosomes to verify the integrity of the contig and the map localization. STS markers are then produced from the end of the inserts of selected cosmids, with the goal being the production of an STS map with markers spaced, on average, every 35-40 kb; however the STSs themselves are not used in the construction of the map. Integration of the map with the genetic map is achieved by hybridization of cloned genes to the arrayed cosmids or by the information provided by the STS markers. The map of the X-chromosome is the most advanced, covering about 62% of the euchromatic portion of the chromosome and ~560 STS markers (Madueno et al., 1995). Maps of the autosomes are anticipated to reach similar coverage within the next few months. Roughly 1300 STS markers with an average length of 400 bp have been determined by cosmid end-sequencing. Eight percent of these STSs have been found to represent either known Drosophila genes or P1 clones and STS markers from the BDGP (providing links to other existing maps) while 3 percent of the STS markers have strong similarities to genes from other organisms, identifying the Drosophila homologues. A cosmid-based mapping effort focused on the small fourth chromosome is being carried out at the University of Alberta, Canada. The euchromatic region of this chromosome contains ~ 70 genes distributed over ~1.2 Mb of DNA. Interestingly, the interspersion pattern of the repeated DNA component of this region is unlike most Drosophila gene-rich regions and more closely resembles the short period interspersion class of repeats found in mammalian DNA. The Alberta group is using a technique they call cross-screening that relies on an array of pairwise cross-hybridization tests performed on a single blot to determine clone overlaps rapidly and that may be particularly well-suited to mapping repeat-rich DNA (Locke et al., 1996). P1-based Maps The BDGP is constructing a map based on P1 clones using a combination of in situ hybridization (Figure 1B) and STS content mapping. The first step was the generation of a framework map based on polytene chromosome in situ hybridization of 2467 P1 clones with an average insert size of 80 kb (Smoller et al., 1991; Hartl et al., 1994). This map provides about 70 percent coverage of the euchromatic genome. The second step of map construction uses STS markers designed from the ends of genomic inserts of individual P1 clones from the framework map. By direct sequencing of the vector/insert junctions of P1 genomic clones, two STS markers per clone can be generated that are separated by the length of the insert, ~80 kb. Over 2300 such P1-end derived STSs have been mapped to date. When the pair of markers is mapped to the library, the resulting contigs extend bi-directionally from the mapping clone and cover on average 200 kb of the genome. This average contig size exceeds that provided by non-random or single-end mapping strategies. Furthermore, computer simulations indicate the number of markers required to assign all clones in the library to contigs will be minimized by using this so-called double-end clone-limited approach (Palazzolo et al. 1991). This phase has been completed; nearly all the 2300 euchromatic P1 clones in the framework map have been assigned to contigs. As of October 1995, 649 contigs cover the genome with an STS localized on average every 50 kb (Kimmerly et al., 1996). The BDGP estimates that about 10% of the euchromatic genome remains unmapped. The position and approximate size of these gaps are known because all contigs have been localized on the polytene chromosome map. Clones not yet assigned to contigs (2200 of the 9216 clones in the 5-hit P1 mapping library) are currently being used as a source of STS markers that will fill in the bulk of the final 10% of the euchromatic genome. Individual clones are selected, mapped by in situ hybridization and used to generate STSs. This phase of the project should be completed during 1996, at which point coverage is expected to be about 98%. Moreover, many of the remaining gaps in the map are likely to be due to overlaps that have not yet been detected rather than to uncloned regions. Subsequent efforts will focus on contig closure, first by screening a larger 10-hit library with contig end probes. This project has provided a critical test of the utility of the P1 cloning system and will result in the first whole genome to be mapped based on a library constructed with large inserts in a vector that is maintained in E. coli as a single copy plasmid. To provide a more direct link between the physical map and the genetic map, the organized set of P1 clones in this physical map are being used as a substrate for further STS content mapping in which the STS sources are derived from markers that are also genetically mapped, including individual genes that have been cloned and sequenced by the research community (350 STSs mapped) and the sites of insertion of P-elements that disrupt essential genes (270 STSs mapped).
 Figure
1. Drosophila chromosomes. (A) Metaphase chromosomes. (B) A portion of a polytene chromosome showing a 4Mb
region from the tip of chromosome arm 3L .The average band visible in polytene chromosome preparations contains about
25 kb of DNA. The bracket indicates the signal obtained by in situ hybridization of an 80 kb P1 clone.
Figure
1. Drosophila chromosomes. (A) Metaphase chromosomes. (B) A portion of a polytene chromosome showing a 4Mb
region from the tip of chromosome arm 3L .The average band visible in polytene chromosome preparations contains about
25 kb of DNA. The bracket indicates the signal obtained by in situ hybridization of an 80 kb P1 clone.
Heterochromatin. One of the most striking and enigmatic aspect of genome organization in multicellular eukaryotes is the division of chromosomes into euchromatic and heterochromatic regions. Heterochromatin is distinguished from euchromatin by its paucity of genes, tightly compacted chromatin structure throughout the cell cycle, unusual staining properties, replication late in S phase, and high content of repetitive sequences. In Drosophila, about one-quarter of the genome is heterochromatic including the centric one-quarter of the X, 2nd and 3rd chromosomes and most of the Y and fourth chromosomes (Gatti and Pimpinelli 1992). Essential functional components are contained within heterochromatic regions, including centromeres, telomeres, ribosomal RNA genes and 30-50 protein coding genes. Progress in understanding the molecular structure and composition of heterochromatin has been limited since much of this DNA, in particular the simple sequence satellite DNA, cannot be stably cloned in existing cosmid, YAC or P1 vectors. However, data obtained while constructing the P1 framework map suggests that the P1 library may contain a substantial proportion of non-satellite heterochromatic sequences (Hartl et al., 1994). Karpen and his collaborators have focused on analyzing the structure and function of heterochromatic regions of the Dp1187 minichromosome, a deletion derivative of the X-chromosome that is only about 1.3 Mb in length (Karpen and Spradling, 1990). Recently, Dp1187 deletion derivatives were generated by irradiation mutagenesis and their structures were determined from PFGE and DNA blot hybridization analyses. Minichromosome derivatives with one break in the euchromatin and one break in the heterochromatin provided single copy entry points for detailed pulse-field restriction mapping of previously inaccessible regions of centric heterochromatin. The map revealed the presence of three large complex "islands" containing middle-repetitive and/or single-copy sequences that are separated by inter-island "seas" of satellite sequences (see Figure 2; Le et al., 1994). Pulsed-field DNA blot analysis demonstrated that in general Drosophila heterochromatin is composed of alternating blocks of complex DNA and simple satellite DNA, each hundreds of kilobases in length. The blocks of complex DNA themselves have considerable substructure and contain many transposable element insertions. The major conclusion from these studies is that a surprising and significant amount of substructure is present deep within Drosophila centric heterochromatin. The presence of repeated DNA has made molecular-genetic analyses of higher eukaryotic centromeres and other heterochromatic inheritance elements extremely difficult. Numerous molecular and cytological studies have associated satellite DNAs with centromeres in mammals, but the exact function of satellite DNAs in mammalian inheritance is unclear in large part because the transmission behavior of molecularly defined components has not been assayed directly. Analyses of the meiotic and mitotic transmission behavior of Dp1187 deletion derivatives has localized sequences necessary for chromosome inheritance within the centric heterochromatin. The essential core of the centromere is contained within a 220 kb region that includes significant amounts of complex DNA (see Figure 2; Murphy and Karpen, 1995).
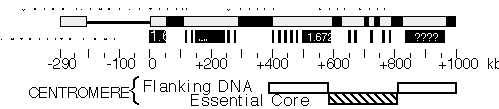
Figure 2. Molecular structure of Dp1187 centric heterochromatin. Sequences to the left of the euchromatin/heterochromatin boundary (position 0 kb) include X-tip euchromatin (solid line) and the subtelomeric heterochromatin (gray box); the 1 Mb of centric heterochromatin is shown as a gray box (0 to +1000 kb). Black boxes are the islands of complex DNA (Tahiti, Moorea, and Bora Bora), which are digested with numerous restriction enzymes that recognize 6 bp sites. Gray boxes in the centric region are blocks that predominantly contain satellite DNA repeats. The approximate locations for some satellites are indicated, where known (1.688=359 bp; 1.672=AATAT). Gray bars within Bora Bora indicate the presence of satellite DNA that separate this island into 4 or more "mini-islands" (Sun and Karpen, unpubl.). The locations of the centromere essential core and redundant flanking regions are indicated below.
Genomic Sequencing The BDGP is currently the only group currently doing production scale Drosophila genomic sequencing. A two-year pilot project involving a sequencing team of seven individuals has just concluded. During this time over 2 Mb of genomic sequence have been completed and deposited in the public databases. The regions sequenced include the Bithorax (~350 kb; Martin et al., 1995) and Antennapedia (~430 kb) homeotic gene complexes, as well as about 1.5 Mb from the 34D-36A genomic region. Although these regions have been heavily studied, their sequences have led to unexpected observations; in the Bithorax complex for example, a glucose transporter gene was discovered in the midst of the complex of homeotic genes. Unlike most genome sequencing projects that employ a shotgun sequencing strategy, the BDGP is using a directed approach to DNA sequencing that has been developed and implemented by the Palazzolo and Martin group at LBNL. This strategy is diagrammed in Figure 3. The approach offers a number of potential advantages: it requires a reduced amount of sequencing; the assembly can be performed in an automated fashion; the assembly is based on redundant information which facilitates the accuracy of the final assembly; and the robust nature of the procedures make them highly amenable to automation. The BDGP was awarded a three year grant from the NCHGR in December 1995 that allows for a four-fold increase over the next 18 months in funding devoted to production sequencing; technological advances and economies of scale should allow a substantially greater relative increase in output. They anticipate that the 120 Mb euchromatic genome can be completed in 5 to 7 years (depending on future funding levels).
 Figure
3. Directed sequencing strategy. The strategy has the following four steps: (1) A P1-based physical map that
provides a set of minimally overlapping clones that represent the genome is generated using an STS content mapping
strategy. (2) DNA from individual P1 clones is sheared to an average size of 3 kb, subcloned into a plasmid vector
and set of minimally overlapping 3 kb subclones is identified. Initial approaches to generating this set of 3 kb
subclones involved using a PCR-based screening method to identify minimally overlapping clones from three dimensional
pools of 960 different clones. Recently, a strategy developed by Bruce Kimmel (unpubl.) has been used in which 192
unique subclones are selected and both ends are then sequenced. The end-specific sequence information is used to
build contigs of 3 kb clones which are used as transposon targets and completely sequenced. Subsequent rounds of
contig-building and subclone sequencing are employed until the 80 kb insert of the P1 is contiguous. (3) gd transposons are mobilized into the 3 kb target sequences by appropriate bacterial matings. Each clone
contains an independent insertion and the insertions in a set of clones are mapped by PCR, using gd element and vector primers. (4) A minimal set of clones with transposons spaced about every 400 bp
(indicated by shaded triangles) are selected and sequenced to give the complete double-stranded sequence of each 3 kb
insert. Binding sites for the sequencing primers are provided by sequences present near the termini of the
transposon.
Figure
3. Directed sequencing strategy. The strategy has the following four steps: (1) A P1-based physical map that
provides a set of minimally overlapping clones that represent the genome is generated using an STS content mapping
strategy. (2) DNA from individual P1 clones is sheared to an average size of 3 kb, subcloned into a plasmid vector
and set of minimally overlapping 3 kb subclones is identified. Initial approaches to generating this set of 3 kb
subclones involved using a PCR-based screening method to identify minimally overlapping clones from three dimensional
pools of 960 different clones. Recently, a strategy developed by Bruce Kimmel (unpubl.) has been used in which 192
unique subclones are selected and both ends are then sequenced. The end-specific sequence information is used to
build contigs of 3 kb clones which are used as transposon targets and completely sequenced. Subsequent rounds of
contig-building and subclone sequencing are employed until the 80 kb insert of the P1 is contiguous. (3) gd transposons are mobilized into the 3 kb target sequences by appropriate bacterial matings. Each clone
contains an independent insertion and the insertions in a set of clones are mapped by PCR, using gd element and vector primers. (4) A minimal set of clones with transposons spaced about every 400 bp
(indicated by shaded triangles) are selected and sequenced to give the complete double-stranded sequence of each 3 kb
insert. Binding sites for the sequencing primers are provided by sequences present near the termini of the
transposon.
Biological Annotation of the Genomic Sequence A key use of the sequence information from the canonical model organisms, such as Drosophila, will be to help interpret the sequence of the human genome. Simply determining the DNA sequence of the genome would be sufficient for comparison with the genomes of other species to identify similarities between genes or protein domains among species. But such similarities are inherently intellectually sterile, unless the biological functions of the genes have been established for one or more of the species being compared. If the model organism genome projects are to be maximally useful in assigning functions to human DNA sequences, they will need to utilize the powerful tools for determining gene function that are available to them so that not only the sequences of the genes, but also their biological functions, are determined. Among the model organisms, Drosophila is particularly well-suited for this role. In terms of evolutionary conservation of sequence similarity, Drosophila is the closest of the invertebrate model organisms to humans (Sidow and Thomas, 1994). Moreover, it terms of morphological, physiological, and behavioral complexity Drosophila is by far the closest to humans of these model organisms, yet its genome is not substantially bigger than the least complex metazoans. Finally, the large Drosophila research community-about one researcher per 2 genes-has provided a wealth of information and understanding unusual in its depth and intellectual breadth. These workers have already extensively characterized about 1000, or 10%, of all Drosophila genes in terms of sequence, gene structure, expression pattern and biological function. Through the efforts of many laboratories over the past 30 years, approximately 25% of the genome has been subjected to saturation mutagenesis experiments which attempt to identify all the genes that can mutate to an easily detectable phenotype. These studies lead to an estimate of 4,000 for the number of genes whose functions are essential for viability, or about one third of the total gene number. One of the best characterized regions is the 1.8 Mb in polytene divisions 34D-36A (Ashburner et al., 1990). The sequence of this region is nearing completion by the BDGP and an attempt is being made to correlate open reading frames and transcription units with genetic loci (see Figure 4). Paul Lasko and Beat Suter, at McGill University, are extending the work of the Wright laboratory (Stathakis et al., 1995) in the genetic and molecular analysis of a similarly sized genomic segment comprising polytene regions 37 and 38 and this will be an early target for the BDGP sequencing efforts. The detailed analysis of these regions-genomic sequence, transcript map, expression data, and mutational analysis-should provide a detailed view of the genomic organization of typical euchromatic regions. Indeed, early attempts at such biological annotation of genomic regions began in the 1980s and provided some of the first indications that the number of transcription units would greatly exceeded the number of genes that could be identified by mutational analysis (Bossy et al., 1984). As part of its efforts to develop and apply tools for large-scale functional analysis, the BDGP has undertaken a novel gene disruption project based on mutagenesis by transposable element insertion (Spradling et al., 1995). Transposable elements provide a powerful tool for correlating genetic and molecular information because they generate a simple, reproducible lesion upon insertion that can be detected much more easily than damage produced by other mutagens. In D. melanogaster the P transposable element has been particularly useful because it moves with high frequency but can be tightly controlled by limiting the availability of an element-encoded transposase. The initial goal of the project is to establish a large collection of Drosophila strains that each contain a single genetically engineered P transposable element insertion that mutates a different gene, in a genome free of other P elements. Moreover, the inserted P elements in BDGP lines carry enhancer-traps that can be used to efficiently acquire information about the expression pattern of disrupted genes. The strains in the current collection disrupt 20-25 percent of essential genes, provide information on their expression patterns, and link the genetic, cytogenetic and physical maps of the Drosophila genome at ~100 kb intervals.

Figure 4. A prototype of the BDGP annotated sequence display. This figure is a screen dump taken from the BDGP's prototype Web genome browser applet, written in Java (G. Helt, unpubl.; for more information contact gregg@fruitfly.berkeley.edu). Correlated genetic map and sequence analysis are shown for a portion of P1 DS02740 (~83kb; GenBank acc. no. L49408). The scale is in kb. Drosophila genes that were previously sequenced by members of the Drosophila research community are shown in black. Lethal P-element insertions whose exact locations were mapped (Spradling et al., 1995) are indicated by the blue-tipped vertical bars. Note that one P element insertion appears to inactivate the gene encoding the L4 ribosomal protein and the other that encoding the ipa-6d homolog. Five Drosophila genes that mutate to detectable phenotypes map between these two P element insertions (J. Roote and M. Ashburner, pers. comm.); three can be assigned to specific transcripts, leaving two complementation groups and five transcripts unassigned. Similiarities of conceptually translated regions to known proteins (BLASTX with a significance cutoff of P=1.0e-8) are shown as red boxes. Results from two gene prediction programs are also shown. Exons predicted by Drosophila GRAIL (Xu et al., 1995) are shown as purple boxes. Exons predicted by a Drosophila-specific version of Genefinder are shown as green boxes. These results were further filtered to eliminate certain gene predictions that were completely "shadowed" by higher-score predictions on the opposite strand. Based on analysis of the database searches and computational gene predictions, primers were designed to probe cDNA libraries for the most likely gene candidates, which are annotated as red arrows. cDNAs were found for all of these candidate transcripts (with the sole exception of the grey arrow on the left; L. Hong, D. Harvey and G. Rubin, unpubl.). These cDNAs are currently being sequenced. The cDNAs are labelled when a significant similarity has been detected by BLASTX: L4 (bacterial ribosomal protein L4, P=4.2e-23); ZK418 (predicted gene from C.elegans, P=6.2e-9); GMF (human glial maturation factor, P=3.0e-43); mkr2 (mouse CNS, P=9.7e-44); ipa-6d (B.subtilus ORF, P=7.2e-12). Significant homologies of the known genes are also worth noting: cactus (human bcl-3 [ikb family], P=2.7e-31); fizzy (human p55cdc, P=5.5e-167); , cornichon (hypothetical yeast protein, P=5.0e-15); SED5 (rat syntaxin, P=9.7e-46).
Databases There are two main databases for Drosophila genome information: FlyBase and the BDGP database. In addition there are numerous specialized databases dealing with many aspects of Drosophila anatomy, gene expression and gene function; a list of these other resources can be found at http://www-leland.stanford.edu/~ger/drosophila.html. FlyBase is the central database for Drosophila genetic information. It captures information from the literature, from the major genome projects and through bulk data provided by sequence and bibliographic data banks. The major genomic data sets in FlyBase include genetic information on genes, alleles, chromosomal aberrations and transposons, as well as molecular information on contigs, chromosomal walks, transcripts and proteins. FlyBase data can be accessed through the World Wide Web (http://morgan.harvard.edu/ or http://www.embl-ebi.ac.uk/flybase/) or by gopher server (flybase.bio.indiana.edu). Data available from the BDGP home page (http://www.fruitfly.org/) include data on the P-element gene-disruption project and monthly updates of the P1-based physical map. A sequence display is under development that presents genomic sequence determined by the BDGP, annotated with the results of homology searches, gene prediction programs and cDNA sequence and expression analyses. A prototype of this display is shown in Figure 4. The BDGP and FlyBase have collaborated to produce the Encyclopaedia of Drosophila, a database and graphical user interface that uses a version of ACEDB (R. Durbin and J. Thierry-Mieg, unpubl.) customized for Drosophila (S. Lewis and C. Harmon, unpubl.) to present an integrated view of much of the BDGP and FlyBase data. The EofD is available as a Macintosh-compatible CD-ROM or by ftp in Macintosh or UNIX versions. ACKNOWLEDGEMENTS I thank my Drosophila colleagues for communicating their results and future plans, and A. Spradling and M. Palazzolo for comments on the manuscript.
REFERENCES Ajioka, J. W., D. A. Smoller, R. W. Jones, J. P. Carulli, A. E. C. Vellek, D. Garza, A. J. Link, I. W. Duncan and D. L. Hartl. 1991. Drosophila genome project: One-hit coverage in yeast artificial chromosomes. Chromosoma 100: 495-509 Ashburner, M., P. Thompson, J. Roote, P. Lasko, Y. Grau, M. El Messal, S. Roth, and P. Simpson. 1990. The genetics of a small autosomal region of Drosophila melanogaster containing the structural gene for alcohol dehydrogenase. Genetics 126: 679-694. Bender, W., P. Spierer, and D. Hogness. 1979. Gene isolation by chromosomal walking. J. Supramol. Struct. 8: 32. Bossy, B., L.M.C. Hall, and P. Spierer. 1984. Genetic activity along 315 kb of the Drosophila chromosome. EMBO J. 3: 2537--2541 Bridges, C. 1937. Correspondence between linkage maps and salivary chromosome structure, as illustrated in the tip of chromosome 2R of Drosophila melanogaster. Cytologia Fujii Jubilee Volume: 745-755 Cai, H., P. Kiefel, J. Yee, and I. Duncan. 1994. A yeast artificial chromosome clone map of the Drosophila genome. Genetics 136: 1385-1401 Garza, D., J. W. Ajioka, D. T. Burke and D. L. Hartl. 1989. Mapping the Drosophila genome with yeast artificial chromosomes. Science 246: 641-646 Gatti, M., and S. Pimpinelli. 1992. Functional elements in Drosophila melanogaster heterochromatin. Annu Rev Genet 26: 239-75 Grunstein, M. and D.S. Hogness, D.S. 1975. Colony hybridization: a method for the isolation of cloned DNAs that contain a specific gene. Proc. natn. Acad. Sci. USA. 72: 3961-3965. Hartl, D. L., D. I. Nurminsky, R.W. Jones, and E. R. Lozovskaya. 1994. Genome structure and evolution in Drosophila: Applications of the framework P1 map. Proc. Natl. Acad. Sci. USA 91: 6824-6829 Karpen, G.H. and A.C. Spradling. 1990. Reduced DNA polytenization of a minichromosome region undergoing position-effect variegation in Drosophila. Cell 63: 97-107. Kimmerly, W., K. Stultz, S. Lewis, K. Lewis, V. Lustre, R. Romero, J. Benke, D. Sun, G. Shirley, C. Martin, M. Palazzolo. 1996. A P1-based physical map of the Drosophila euchromatic genome, submitted. Le, M.-H., D. Duricka, and G.H. Karpen. 1995. Islands of complex DNA are widespread in Drosophila melanogaster centric heterochromatin. Genetics 141: 283-303 Locke J., G. Rairdan, H. McDermid, D. Nash, D. Pilgrim, J. Bell, K. Roy and R. Hodgetts. 1996. Cross-screening: a new method to assemble clones rapidly and unambiguously into contigs. (submitted). Madueno, E., G. Papagiannakis, G.A. Rimmington, R.D.C. Saunders, C. Savakis, I. Siden-Kiamos, G. Skavdis, L. Spanos, J. Trenear, P. Adam, M. Ashburner, P. Benos, V.N. Bolshakov, D. Coulson, D.M. Glover, S. Herrmann, F.C. Kafatos, C. Louis, T. Majerus, J. Modolell. 1995. A physical map of the X chromosome of Drosophila melanogaster: Cosmid contigs and sequence tagged sites. Genetics 139: 1631--1647 Martin, C.H., C.A. Mayeda, C.A. Davis, C.L. Ericsson, J.D. Knafels, D.R. Mathog, S.E. Celniker, E.B. Lewis, and M.J. Palazzolo. 1995. Complete sequence of the bithorax complex of Drosophila. Proc. Natl. Acad. Sci. USA 92: 8398-8402 Murphy, T. and G.H. Karpen. 1995. Localization of centromere function in a Drosophila minichromosome. Cell 82: 599-609. Palazzolo, M.J., S.A. Sawyer, C.H. Martin, D.A. Smoller, and D.L. Hartl. 1991. Optimized strategies for STS selection in genome mapping. Proc. Natl. Acad. Sci. 88: 8034-8038 Pardue, M.L., S.A. Gerbi, R.A. Eckhardt, and J.G. Gall. 1970. Cytological localization of DNA complementary to ribosomal RNA in polytene chromosomes of Diptera. Chromosoma 29: 268--290 Rubin, G.M., D.J. Finnegan, and D.S. Hogness. 1976. The chromosomal arrangement of coding sequences in a family of repeated genes. Prog. Nucleic Acid Res. Molec. Biol. 19:;221-226. Siden-Kiamos I., R.D.C. Saunders, L. Spanos, T. Majerus. J. Trenear, C. Savakis, C. Louis., D.M. Glover, M. Ashburner, F.C. Kafatos. 1990. Towards a physical map of the Drosophila melanogaster genome: mapping of cosmid clones within defined genomic divisions. Nucleic Acids Res. 18: 6261--6270 Sidow, A. and W.K. Thomas. 1994. A molecular evolutionary framwork for eukaryonic model organisms. Curr. Biol. 4: 596-603. Smoller, D. A., D. Petrov and D. L. Hartl. 1991. Characterization of bacteriophage P1 library containing inserts of Drosophila DNA of 75-100 kilobase pairs. Chromosoma 100: 487-494. Spradling, A. C., D. M. Stern, I. Kiss, J. Roote, T. Laverty and G.M. Rubin. 1995. Gene disruptions using P transposable elements: An integral component of the Drosophila genome project. Proc. Natl. Acad. Sci. USA. 92: 10824-10830 Stathakis, D. G., E. S. Pentz, M. E. Freeman, J. Kullman, G. R. Hankins, N. J. Pearlson and T. R. F. Wright. 1995. The genetic and molecular organization of the Dopa decarboxylase gene cluster of Drosophila melanogaster. Genetics 141: 629-655 Sturtevant, A. H. 1913. The linear arrangement of six sex-linked factors in Drosophila, as shown by their mode of association. J. Exp. Zool. 14: 43-59 Wensink, P.C., D.J. Finnegan, J.E. Donelson, and D.S. Hogness. 1974. A system for mapping DNA sequences in the chromosomes of Drosophila melanogaster. Cell. 3: 315-325. Xu, Y., G. Helt, J.R.Einstein, G.Rubin, and E.C.Uberbacher. 1995. Drosophila GRAIL: An intelligent system for gene recognition in Drosophila DNA sequences..Proc. First Int. Symp. on Intelligence in Neural and Biological Systems. 128-135.