Projects
Genomic DNA Sequencing Project FAQ
How big is the Drosophila genome? How many genes does it have?
How much of the genome sequence is in GenBank?
How was the sequencing done? Are there still gaps?
What is the difference between Release 1, 2, and 3?
How were the annotations done? How reliable are they?
How do I view the annotations?
How do I find the sequence of a particular annotation?
How do I report errors in the annotations or in the genomic sequence?
How were map positions determined for the annotations?
Should I trust the sequence of the transposons in Release 1 and 2?
How was the BAC physical map made?
Why should I use BAC clones instead of P1 clones?
How do I find a BAC for my region?
How do I request a BAC clone? How do I get a copy of the BAC library?
How were the genomic libraries made?
How do I download all of the genomic sequence, or other databases?
When is the Drosophila pseudoobscura genome going to be sequenced, and by whom?
How big is the Drosophila genome? How many genes does it have?
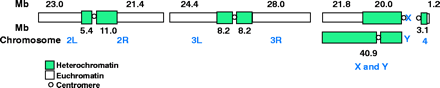
The genome sequence (Release 1.0) is described in Adams et al. 2000. The genome is estimated to be 180 Mb: 120 Mb of euchromatin and 60 Mb of unclonable heterochromatin. Scaffolds containing 116.2 Mb of sequence from the whole genome shotgun sequence map to chromosome arms, and scaffolds containing 3.8 Mb are unmapped. Some of the latter belong to the Y chromosome. The approximate sizes of the chromosome arms were determined by summing the sequence and the estimated gap sizes.
Fig. 1, Adams et al. 2000
As presently annotated, the Drosophila genome has 13,601 genes. This number will undoubtedly change as the genome is re-annotated.
How much of the genome sequence is in GenBank?
GenBank contains the major genome sequence scaffolds described in Adams et al. 2000 (accession nos. AE002566 - AE003403). This assembly totals 116.2 Mb after initial gap closure. Fifty major scaffolds containing 114.8 Mb were mapped to the chromosomes; 704 scaffolds covering 3.8 Mb remain unmapped. The major scaffolds were cut into ~350 kb pieces, and each piece has its own accession number.
How was the sequencing done? Are there still gaps?
The genome sequence was determined by a whole genome shotgun sequencing strategy supported by clone-based sequencing and a BAC physical map (Adams et al. 2000; Myers et al. 2000; Hoskins et al. 2000). The sequence data set consists of (1) over 3 million sequence reads generated from the ends of 2 kb and 10 kb genomic clones (Celera Genomics); (2) 19,738 sequence reads from the ends of BAC clones (Genoscope); (3) 29 Mb of finished sequence from BAC, P1 and cosmid clones (BDGP-LBNL and EDGP); and (4) draft sequence reads (>= 1.5 X coverage) from 825 BAC and P1 clones spanning 97% of the euchromatic portion of the genome (BDGP sites at LBNL and Baylor College of Medicine). Paired-end sequence data was essential to the correct assembly of the whole genome shotgun data at Celera Genomics; 72% of sequence reads in the whole genome shotgun were in the form of paired-end sequence and provided the information necessary to assemble ordered and oriented sequence contigs.
Finishing is being completed by the BDGP sites at LBNL and Baylor College of Medicine in collaboration with Celera Genomics. The whole genome shotgun sequence trace data generated at Celera and the clone-based sequence traces generated by the BDGP are being re-assembled using Phrap at LBNL and Baylor in a series of BAC-based assemblies. All gaps are being filled, and low-quality sequence is being brought to high quality. A variety of methods are being used including sequencing BAC templates with custom primers, generating and sequencing PCR products, and identifying plasmid clones for transposon-based sequencing.
Most gaps result from repeat sequences, which were masked for the whole genome shotgun sequence assembly. One type results primarily from retrotransposons. The retrotransposon gaps are generally large, on the order of 4 to 8 kb. The other type of gap is small and results from homopolymeric and dinucleotide repeat sequences. We are working to close all gaps and resolve all ambiguities in the genome sequence assembly.
What is the difference between Release 1 and Release 2?
Please see our Release 2 and Release 3 Notes.
How were the annotations done? How reliable are they?
The current annotation of the genome sequence is described in Adams et al. 2000 and Rubin et al. 2000
The sequence was annotated at Celera at an 'Annotation Jamboree' using gene prediction programs and limited human curation. Over the next six months to one year, the BDGP will finish the sequence to the Phase III standard. As finishing is completed for an interval, that interval will be re-annotated; when an entire arm is finished and re-annotated, there will be a new release of that arm. Between now and then, we need your help! You will undoubtedly notice errors in the sequence and mistakes in the annotations. Help us to correct them by sending us an Error Report. We will post your comments as additions to the gene annotation record and, when we reach that part of the genome, we will use them as an aid in the finishing process and to correct gene annotations.
Treat the current annotations with skepticism. The annotations were done using a combination of gene prediction programs and limited human curation. Gene prediction programs do a very good job of identifying exons, but are less proficient at determining exact splice sites. It is likely that only a minority of the pr edicted gene structures in the current annotated set are completely correct. Another common problem is that two genes are merged into one, or conversely, that one gene has been split into two. If you know that a particular annotation is incorrect, please help us by filling out an Error Report form.
The functional classifications that you see in Gadfly were done computationally as a way to manage the task of annotation and have had limited human oversight; therefore some of the classifications of a predicted protein's function may be wrong. You must not unquestioningly accept them.
The BLAST searches reported in Gadfly were run in November 1999. You must do your own BLAST searches in order to get the most current results. When we re-annotate the genome, we will re-run BLAST, but until the annotations are refined, it is important to do your own BLAST searches.
How do I view the annotations?
From either this site or FlyBase, follow the links to Gadfly, the Genome Annotation Database. If you search using a gene name, symbol, or CG/CT/FBan number, you will be able to view the annotation of that gene along with surrounding genes.
How do I find the sequence of a particular annotation?
In Gadfly, above the annotation itself, is a menu whose default is "Display full report". In that menu are two options for the cDNA FASTA sequence and translation FASTA sequence. We hope to have available soon the genomic sequence of the genes in Gadfly also.
How do I report errors in the annotations or in the genomic sequence?
In Gadfly, to the right of the annotation, is a button that says "Fix this annotation". Clicking on this takes you to an error report form which you can use to correct the annotations. These become part of the gene record, and will be valuable to us as we reannotate the genome.
Changes to the genomic sequence that you believe are not due to polymorphisms between your strain and the sequenced isogenic y; cn bw sp strain can be reported [HERE].
How were map positions determined for the annotations?
All the predicted genes have now been incorporated into FlyBase with inferred cytology. The inference system we have used is to take the estimates which Sorsa published a few years ago of the size in kb of each polytene band. These estimates can be summed to give the length (according to Sorsa) in kb of a region between two very well-mapped genes ("anchors") that are also identified on the genome. The genome sequence gives a different number for that length, of course. So we then just applied a scaling factor, i.e. we calculated the cytology of each genome gene in the region between the anchors by interpolation from its sequence coordinates. The anchors we chose are spaced about a number division apart. The scaling works out slightly different for each inter-anchor region, of course, and we reckon that even in the middle of a region the error should never be more than a couple of bands.
The dataset is based on the genome coordinates, which includes only the assemblable and mappable euchromatic sequence. This means that the first few polytene bands of most arms actually have negative sequence coordinates, indicating that those bands are beyond the end of the mapped genome sequence, and conversely some arms start at numbers greater than 0 indicating that the sequence extends into the region beyond polytene bands. As the remaining gaps in the genome sequence are filled, some currently unmappable stretches of sequence (especially near centromeres) will be joined up with the main sequence, and that will shift all the coordinates. Smaller changes will occur as a result of other gap-filling in the middle of arms. These will be reflected in updates.
Should I trust the sequence of the transposons in Release 1 and 2?
No. As a result of the whole genome shotgun assembly, the sequence of each transposon in Releases 1 and 2 is a composite derived from a number of elements of that transposon type. The extent of the composite varies among the transposons depending on the length of the traces that run from unique sequence into the transposon. The sequence is most often not the actual sequence of the particular transposon at that location. Users are warned not to base too much on any analysis of these transposable element sequences.
As we finish the sequence to high quality, we are replacing these composite sequences with the actual sequences present at each location in the y; cn bw sp strain. The BAC sequence submitted to GenBank and Release 3 contain transposons that have been resequenced and the sequence relects the sequence of the transposon at that location.
How was the BAC physical map made?
- The BAC map of chromosomes 2 & 3 is described in Hoskins et al. 2000 and can be viewed using our Java map viewer ArmView.
- A BAC map of the X chromosome produced by the EDGP can be viewed [HERE].
- A BAC map of chromosome 4 produced in the laboratory of Ross Hodgetts is described in Locke et al. 2000.
To construct the BAC map of chromosomes 2 and 3, we used STS markers derived from the P1 map and 690 new STSs designed from the ends of the BAC clones themselves. The end sequences of over 12,000 of these BACs were determined in collaboration with Genoscope. These sequences are available in the EMBL and GenBank sequence databases; the Berkeley Fly database HTML reports for individual BAC clones also show these sequences when available.
We carried out STS content mapping by filter hybridization to high-density arrays representing the entire RPCI-98 BAC library. These data were assembled using SEGMAP and manually edited. The physical map of chromosomes 2 & 3 has been essentially completed, and can be viewed using our Java map viewer ArmView. The BAC map was used to select a tiling path of approximately 1000 BAC clones for draft sequencing and sequence finishing. This tiling path can be viewed on the Release 2.5 Genomic Sequence pages. Most of these BAC clones have been localized by in situ hybridization to polytene chromosomes by the BDGP Cytogenetics Core, and the remainder will be over the next year. 10,000 of the clones in the BAC library (approximately 10-hit) have been restriction enzyme fingerprinted with EcoRI at the BDGP's Baylor College of Medicine site.
Why should I use BAC clones instead of P1 clones?
DNA samples for construction of the BAC library and WGS plasmid libraries were prepared from y; cn bw sp at about the same time in the summer of 1998. The Release 1.0 genome sequence derives from these libraries. The P1 library was constructed in 1991 (Smoller et al. 1991), and the y; cn bw sp strain evolved over those years. The primary difference between the BAC and whole genome shotgun libraries and the P1 library is the location of retrotransposons.
The BAC based physical map is more complete and accurate than the P1 map and has been verified by comparison to the whole genome shotgun sequence assembly. As the genome sequence is finished, sequence assemblies are being verified by comparison to BAC restriction fingerprints.
Many of the P1 clones do not yield intact DNA in standard preps, due to the host strain genotype. Preparation of BAC DNA is much more reliable. Protocols for preparations of BAC DNA are available on the BACPAC Resources page.
As the P1 library is largely obsolete, P1 clones are no longer available from the distribution centers. All requests for P1 clones should be directed to the BDGP at bdgp@fruitfly.org; the BDGP will distribute a P1 clone only if there is a compelling scientific reason that a corresponding BAC is not sufficient. We thank the 13 laboratories that volunteered to distribute P1 clones during the 90's for their generous service.
How do I find a BAC for my region?
We are currently in the process of improving the web-based interfaces and the BDGP databases. In the future, it will be easier to identify BACs from a particular region of interest. For the time being, the best available routes to selecting a BAC are
-
By cytology: Using ArmView2. Click on the arm of interest, then click on a pink BAC clone in the cytological area of interest. This will take you to our Genome browser, and you can click on individual genes in blue to get the approximate cytology, or scroll to the bottom of the page for in situ images of BACs in this area. The BAC will appear as an arrow at the top of the display. You can scroll to the left or right, or zoom in or out.
-
By sequence: Using Fly BLAST Select "BDGP/EDGP genomic clones". In the results list, if you click on the accession number, it will take you to the GenBank entry for the sequence of that clone.
-
By clone ID: Go to the Genome browser. Enter "Clone:BACR31D05" for example, it will take you to a map of that clone.
-
BAC-based STS content maps:
Once you find BAC clones over your region of interest, we recommend selecting th ree or so, and restriction mapping them to make sure that the correct clones wer e shipped and that they overlap your genomic region.
How do I request a BAC clone? How do I get a copy of the BAC library?
Information about obtaining BAC clones, BAC libraries and nylon filters representing the BAC libraries can be found on our page BDGP Resources - Materials .
Please note that the BDGP and BACPAC Resource Center have slightly different nomenclature systems for the BAC clones. A clone called "BACR48M07" by the BDGP should be referred to as "RPCI-98 48.M.7" in communications with BACPAC. In both systems, the first number refers to a library microtiter plate, and the letter and second number refer to the row and column in a 384-well plate format. The BDGP names are "zero-padded"; in the BACPAC names, plate, row and column are separated by periods. We apologize for any confusion arising from the two parallel naming conventions; they exist because we wanted BDGP clone names (BACR for EcoRI) to follow the nomenclature already established for two BAC libraries produced by the European Drosophila Genome Project, BACH (HinDIII) and BACN (NdeI)."
How were the genomic libraries made?
The BDGP Drosophila melanogaster BAC library was prepared by Kazutoyo Osoegawa and Aaron Mammoser in Pieter de Jong's laboratory in the Department of Cancer Genetics at the Roswell Park Cancer Institute in Buffalo, NY. The library is named "RPCI-98" and was constructed by partial EcoRI digestion of Drosophila DNA provided by the BDGP from the isogenic strain y2; cn bw sp, the same strain used for the P1 and cDNA libraries.
The construction of the RPCI-98 BAC library is described at http://www.chori.org/bacpac/98framedromel.htm.
The construction of the DrosBAC library is described at http://www.hgmp.mrc.ac.uk/Biology/descriptions/dros_bac.html
An explanation of the construction of the bacteriophage P1 library, can be found on the Construction of the P1-based framework page.
How do I download all of the genomic sequence, or other databases?
Click on the Download link from the left hand menu on the BDGP home page. Descriptions are provided for all of the available databases.
When is the Drosophila pseudoobscura genome going to be sequenced, and by whom?
Please read more about this at the Baylor College of Medicine Human Genome Sequencing Center.